The actions of cyclin D-dependent kinases serve to combine extracellular signing throughout G1 section with the cell-cycle engine that regulates DNA replication and mitosis. Induction of D-type cyclins and their meeting into holoenzyme complexes rely upon mitogen stimulation.
Conversely, the truth that D-type cyclins are labile proteins ensures that the subunit pool shrinks quickly when cells are disadvantaged of mitogens. Phosphorylation of cyclin D1 on a single threonine residue close to the carboxyl terminus (Thr-286) positively regulates proteasomal degradation of D1.
Now, we display that glycogen synthase kinase-3beta (GSK-3beta) phosphorylates cyclin D1 particularly on Thr-286, thereby triggering fast cyclin D1 turnover. Because the exercise of GSK-3beta will be inhibited by signing by means of a pathway that sequentially includes Ras, phosphatidylinositol-3-OH kinase (PI3K), and protein kinase B (Akt), the turnover of cyclin D1, like its meeting, can be Ras dependent and, therefore, mitogen regulated.
In distinction, Ras mutants faulty in PI3K signing, or constitutively energetic mitogen-activated protein kinase–kinase (MEK1) mutants that act downstream of Ras to activate extracellular sign–regulated protein kinases (ERKs), can not stabilize cyclin D1.
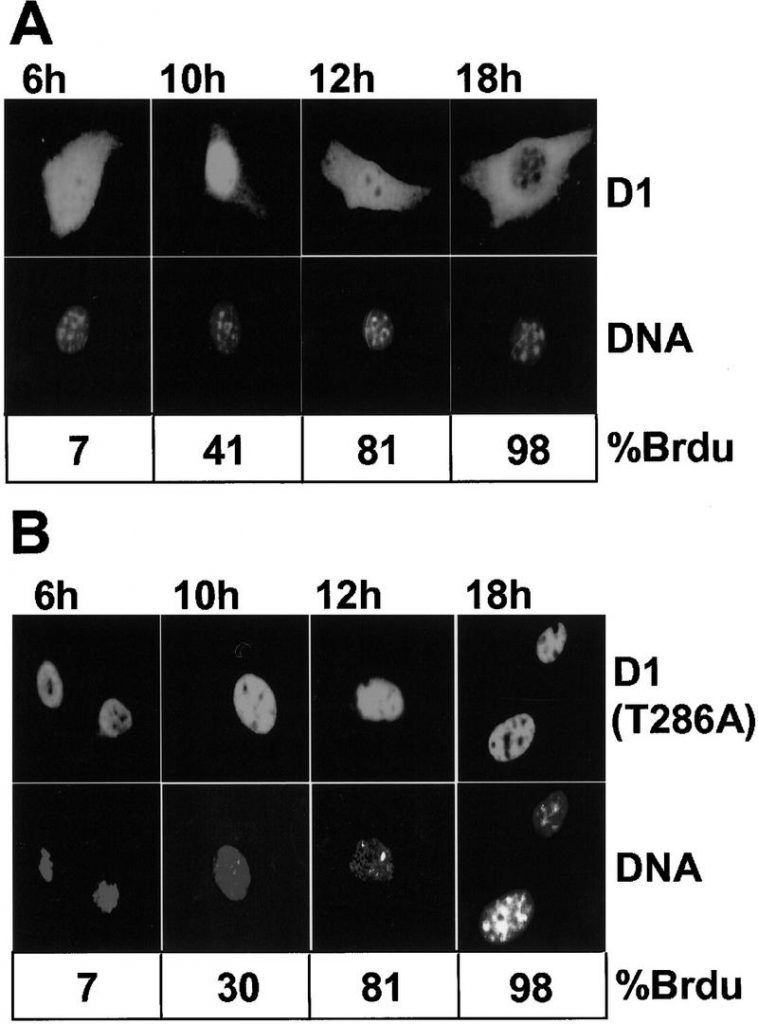
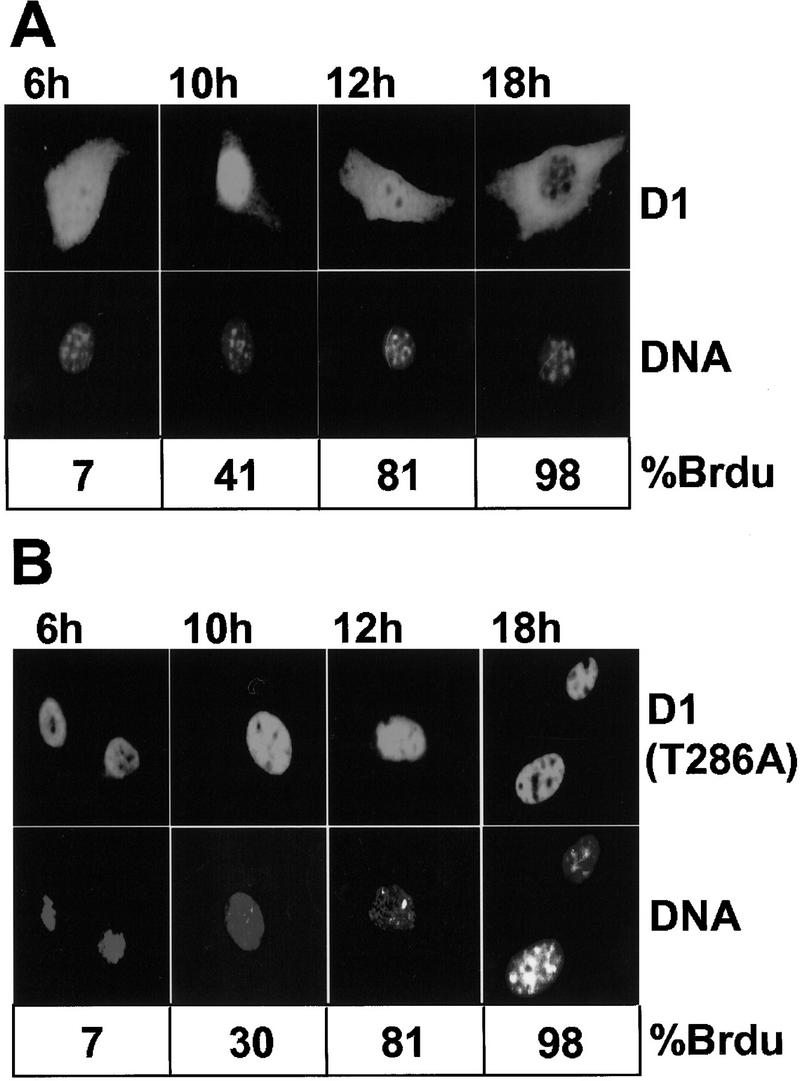
In direct distinction to cyclin D1, which accumulates within the nucleus throughout G1 section and exits into the cytoplasm throughout S section, GSK-3beta is predominantly cytoplasmic throughout G1 section, however a major fraction enters the nucleus throughout S section.
A extremely secure D1 mutant during which an alanine is substituted for the threonine at place 286 and that’s refractory to phosphorylation by GSK-3beta remained within the nucleus all through the cell cycle. Overexpression of an energetic, however not a kinase-defective, type of GSK-3beta in mouse fibroblasts brought on a redistribution of cyclin D1 from the cell nucleus to the cytoplasm.
Therefore, phosphorylation and proteolytic turnover of cyclin D1 and its subcellular localization through the cell division cycle are linked by means of the motion of GSK-3beta.
Autophagy is a mobile catabolic course of that depends on the cooperation of autophagosomes and lysosomes. During hunger, the cell expands each compartments to boost degradation processes.
We discovered that hunger prompts a transcriptional program that controls main steps of the autophagic pathway, together with autophagosome formation, autophagosome-lysosome fusion, and substrate degradation. The transcription issue EB (TFEB), a grasp gene for lysosomal biogenesis, coordinated this program by driving expression of autophagy and lysosomal genes.
Nuclear localization and exercise of TFEB have been regulated by serine phosphorylation mediated by the extracellular sign–regulated kinase 2, whose exercise was tuned by the degrees of extracellular vitamins. Thus, a mitogen-activated protein kinase-dependent mechanism regulates autophagy by controlling the biogenesis and partnership of two distinct mobile organelles.
